Энтропия
Уравнение Клаузиуса (31)
можно обобщить на случай, когда система, совершающая циклический процесс,
обменивается теплотой не с двумя, а с
|
(34) |
где
|
(35) |
Уравнение Клаузиуса справедливо, если все части цикла представляют собой обратимые процессы и цикл является обратимым. Если же цикл включает в себя необратимый процесс и оказывается необратимым, то уравнение (35) не выполняется и имеет место неравенство Клаузиуса
|
(36) |
Свойство обратимых циклов, выражаемое уравнением
Клаузиуса (35),
можно представить в иной форме. Рассмотрим обратимый процесс, при котором
система переходит из состояния
|
|

На рис. 1.4 показаны два таких процесса. Рассмотрим цикл
|
(37) |
Разобьем интеграл по циклу на сумму двух интегралов,
первый из которых берется по пути
|
|
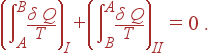
Если перенести теперь второй интеграл направо и поменять в нем пределы интегрирования, то получается соотношение
|
|
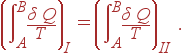
Таким образом, интеграл
|
|
зависит только от начального и конечного состояний
Это свойство позволяет ввести новую функцию состояния системы, называемую энтропией,
которая представляет собой исключительно важную термодинамическую величину.
Энтропией состояния
|
(38) |
где интеграл берется по произвольному обратимому
процессу от состояния
Определим разность энтропий в состояниях
|
|
Разобьем интеграл по циклу на сумму трех интегралов:
|
|
Согласно определению (38)
первый интеграл представляет энтропию в состоянии
|
(39) |
где интеграл берется по произвольному обратимому
процессу от
Формула (38)
представляет энтропию с точностью до аддитивной постоянной. Эта
неопределенность не проявляется, если нас интересует разность энтропии в
различных состояниях (39),
которая не зависит от состояния
Если от конечного процесса перейти к бесконечно малому процессу, то (39) принимает вид
|
(40) |
т.е. изменение энтропии при бесконечно малом
обратимом процессе равно количеству теплоты
Формула (40)
означает, что отношение
Подставим сооотношение
|
(41) |
которое называется основным термодинамическим тождеством. Оно объединяет первый и второй законы термодинамики и справедливо для бесконечно малых обратимых процессов.
Энтропия составной системы равна сумме энтропий ее частей. Это верно, если энергия и работа аддитивны, т.е. если энергия системы равна сумме энергий всех ее частей, а работа, совершаемая системой, равна сумме работ, совершаемых ее частями. Энергия системы аддитивна, если можно пренебречь энергией взаимодействия подсистем по сравнению с их энергиями, т.е. если поверхностная энергия мала по сравнению с объемной. Для системы, состоящей из двух различных веществ, это так, если они не очень сильно размельчены. В противном случае поверхностная энергия играет важную роль.
Итак, если система состоит из двух частей и внутренняя энергия и работа аддитивны,
|
|
то поглощаемая системой теплота также аддитивна:
|
(42) |
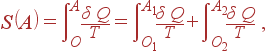
где
Отметим, что выражение (42) можно использовать для определения энтропии системы, не находящейся в состоянии равновесия, если ее можно разбить на части, находящиеся в равновесных состояниях.
Изменение энтропии системы при переходе из состояния
|
|

Применяя неравенство Клаузиуса (36) к этому циклу, имеем
|
(43) |
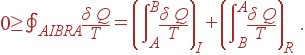
К обратимому процессу ![]() Это приводит к неравенству
Это приводит к неравенству
|
(44) |
где интеграл берется по произвольному необратимому
процессу
Закон возрастания энтропии
Применим неравенство (44)
к изолированной системе, которая никак не взаимодействует с окружающей
средой. Поскольку для такой системы
|
(45) |
т.е. для любого процесса в изолированной системе энтропия конечного состояния не может быть меньше энтрпии начального состояния. Это – закон возрастания энтропии.
Если процесс обратим, то в (45) стоит знак равенства, т.е. энтропия системы не меняется.
Закон возрастания энтропии справедлив только для изолированных систем. С помощью внешней системы можно уменьшить энтропию тела. Однако суммарная энтропия тела и внешней системы уменьшиться не может.
Если изолированная система находится в состоянии с максимальной энтропией, сооответствующей ее энергии, то в ней не могут происходить никакие процессы, поскольку любой процесс привел бы к уменьшению энтропии. Таким образом, состояние с максимальной энтропией является наиболее устойчивым состоянием изолированной системы.
Самопроизвольные процессы в изолированных системах идут в направлении роста энтропии. Продемонстрируем это двумя примерами.
1. Рассмотрим теплообмен между двумя частями системы
|
|
При этом энтропия тела
|
(46) |
Поскольку
2. Рассмотрим теперь выделение теплоты при трении. Энтропия тела, которое нагревается при трении, возрастает. Это увеличение энтропии не компенсируется уменьшением энтропии других частей системы, так как теплота получена из работы.
Энтропия равновесных систем
Рассмотрим систему, состояние которой в равновесии определяется какими-либо
двумя из трех переменных
|
(47) |
Важное отличие (47)
от (13)
состоит в том, что ![]() и, следовательно,
одинаковы. В противоположность этому не существует функции состояния
и, следовательно,
одинаковы. В противоположность этому не существует функции состояния ![]() , полным дифференциалом которой была бы
правая часть (13).
Действительно, количество теплоты
, полным дифференциалом которой была бы
правая часть (13).
Действительно, количество теплоты
|
(48) |
где
|
(49) |
Но разность ![]() и,
следовательно,
и,
следовательно,
Поскольку
|
|
Сравнение этого выражения с (47) дает уравнения
|
(50) |
Если первое уравнение продифференцировать по
|
(51) |
Производя дифференцирование по
|
|
откуда
|
(52) |
Это – важное соотношение, которое часто используется при решении термодинамических задач. В частности, с его помощью можно показать, что внутренняя энергия идеального газа не зависит от объема. Действительно, используя уравнение состояния идеального газа (3), имеем
|
(53) |
так что (52) дает
|
|
т.е. внутренняя энергия идеального газа зависит только от температуры:
|
(54) |
Выражение для частной производной
|
|
Подставляя это выражение в формулу (47)
для
|
(55) |
В качестве примера вычислим энтропию одного моля
идеального газа. Принимая во внимание, что ![]() и
выражая давление
и
выражая давление ![]() из
уравнения состояния, преобразуем (55)
к виду
из
уравнения состояния, преобразуем (55)
к виду
|
(56) |
Это выражение удовлетворяет основному требованию,
предъявляемому к полным дифференциалам, а именно: перекрестные производные от
функций, на которые умножаются ![]() равны друг другу. Интегрируя (56),
находим
равны друг другу. Интегрируя (56),
находим
|
(57) |
где
Иначе обстоит дело с выражением для элементарной теплоты
|
(58) |
В этом случае перекрестные производные не равны друг
другу. Поэтому функции ![]() полным дифференциалом которой было бы это
выражение, не существует.
полным дифференциалом которой было бы это
выражение, не существует.
В заключение сформулируем общий критерий, который позволяет заключить, является ли то или иное выражение полным дифференциалом. Дифференциальная форма
|
(59) |
является полным дифференциалом некоторой функции
|
(60) |
Докажем это. Полный дифференциал
|
(61) |
Сравнение (59) с (61) приводит к уравнениям
|
(62) |
Дифференцируя теперь первое из этих уравнений по
|
|
мы, действительно, приходим к условию (60).
Третий закон термодинамики (теорема Нернста)
В разделе 1.11
мы уже упоминали, что определение энтропии (38)
не является однозначным, поскольку произвол в выборе начала отсчета
Значение аддитивной константы, возникающей при определении энтропии, устанавливается теоремой Нернста, которую часто называют третьим законом термодинамики: энтропия любой системы при абсолютном нуле температуры всегда может быть принята равной нулю.
Физический смысл теоремы состоит в том, что при
|
(63) |
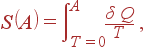
где интегрирование производится вдоль обратимого процесса,
начинающегося от состояния при
В термодинамике теорема Нернста принимается как постулат. Доказывается она методами квантовой статистики.
Из теоремы Нернста следует важный вывод о поведении теплоемкости тел при
|
(64) |
где
|
(65) |
Из этой формулы видно, что если бы теплоемкость тела
при абсолютном нуле,
|
(66) |
Этот вывод находится в согласии с экспериментальными
данными по теплоемкости тел при
Следет отметить, что (66) относится не только к твердым телам, но и к газам. Сделанное ранее утверждение о том, что теплоемкость идеального газа не зависит от температуры, справедливо только для не слишком низких температур. При этом нужно иметь в виду два обстоятельства.
1. При низких температурах свойства любого газа сильно отличаются от свойств идеального газа, т.е. вблизи абсолютного нуля ни одно вещество не является идеальным газом.
2. Если бы даже идеальный газ мог существовать вблизи нуля температуры,
то строгое вычисление его теплоемкости методами квантовой статистики
показывает, что она стремилась бы к нулю при